Partenaires



Accueil du site > Production scientifique > Evaluation of Ca2+ Binding Sites in Tacrolimus by Infrared Multiple Photon Dissociation Spectroscopy
Evaluation of Ca2+ Binding Sites in Tacrolimus by Infrared Multiple Photon Dissociation Spectroscopy
Date de publication: 1er novembre 2018
M. Masson ; R. Karpfenstein ; D. de Oliveira-Silva ; J.-M. Teuler ; P. Archirel ; P. Maître ; T. C. Correra
J. Phys. Chem. B. 122 9860-9868 (2018). DOI
Travail réalisé sur le site de l’Université Paris Sud.
Abstract
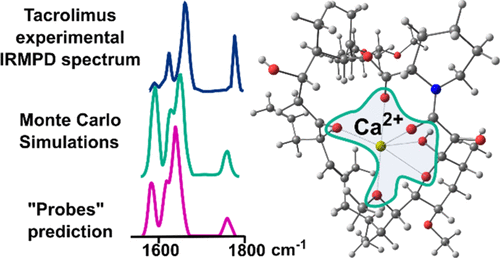
Tacrolimus (TAC) is an efficient immunosuppressant used in organ transplantation procedures. There is an intrinsic correlation between TAC and Ca2+ because of the dependence of its action mechanism on calcium and calcineurin, and the role of ion coordination on TAC identification and quantitation. To depict the Ca2+ binding sites in TAC, this work carried out gas-phase vibrational infrared multiple photon dissociation spectroscopy of [Ca(TAC)](2+) and of three other TAC mimetic molecules (probes 1-3). Density functional theory (DFT) and Monte Carlo (MC) simulations were also used to support the experimental data assignment, and natural bond orbital (NBO) analysis was carried out to depict the coordination sphere. PM3 and B3LYP/6-31G(d) levels of theory displayed similar trends during the MC simulations, suggesting that PM3 is a viable alternative to more expensive DFT calculations, at least during the conformational analysis step. Infrared spectroscopy of the [Ca(probeX)(1)](2+) and [Ca(probeX)(3)](2+) (X = 1-3) complexes allowed for a useful guide for building guess geometries and for the band assignment of the [Ca(TAC)](2+) complex. Nevertheless, the MC approach was particularly useful for exploring the potential energy surface. The lowest energy conformation for [Ca(TAC)](2+) was found by MC simulations and is 32.92 kJ mol(-1) lower in energy than the one found by comparing the results obtained for Ca2+ coordination in probes, despite the calculated spectra being virtually identical. Both approaches are good ways to depict the coordination sites, and these results suggest that using small molecules as models is a reliable approach to depict the geometry or coordination sites of extensive ions, yielding a robust correlation between experimental and theoretical spectra. Furthermore, MC survey produced a lower energy conformation with a good match to the experimental results. Both methods depict the Ca2+ coordination sphere as a hexacoordinated environment where the main coordination centers are carbonyl groups.